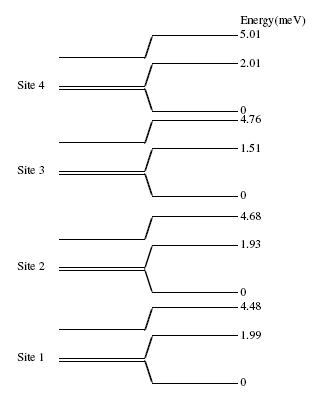
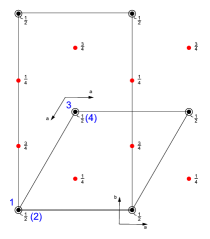
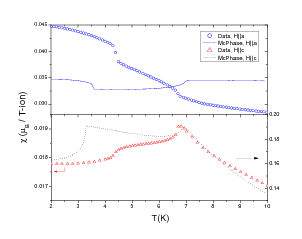
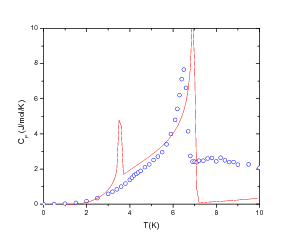
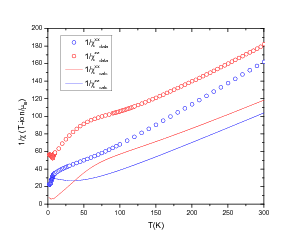
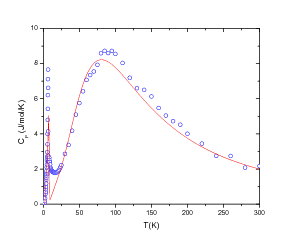
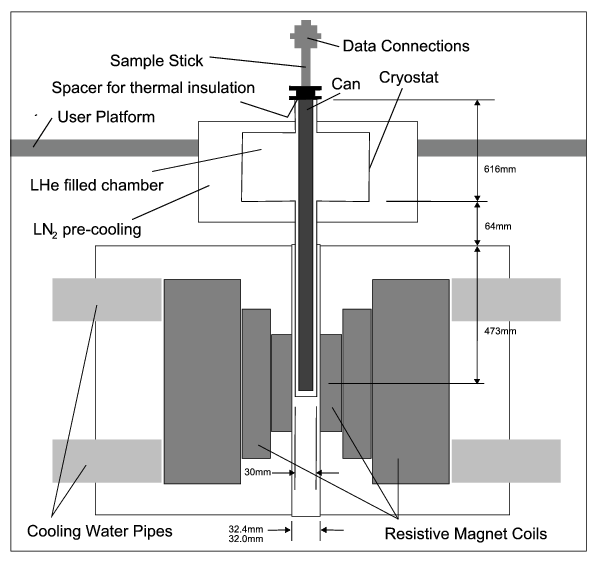
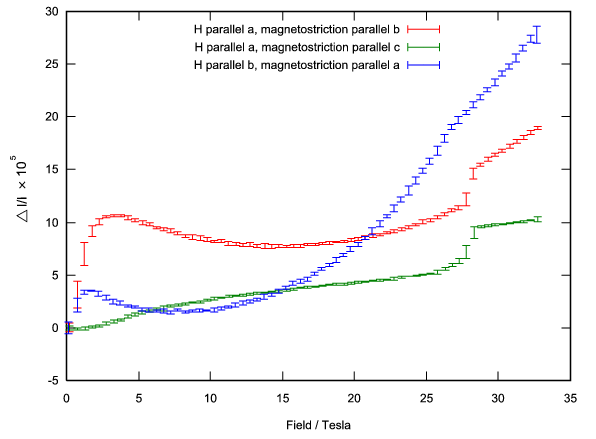
function [FitB2,FitB4,FitB6] = fitengy(A,B2,B4,B6,E,ind_par,constraints)
% fitengy(A,B2,B4,B6) - attempts to fit crystal field parameters in meV.
%
% Syntax: [FitB2,FitB4,FitB6] = fitengy(A,B2,B4,B6,E,ind_par,constraints)
%
% Inputs: A = [L S J] with L,S,J being the angular momentum quantum
% numbers of the ground state multiplet;
% -2 -1 0 1 2
% B2 = [B B B B B ]
% 2 2 2 2 2 are guesses for the crystal field
% B4 = [B_4^{-4} ... B_4^4] parameters in Stevens normalisation.
% B6 = [B_6^{-6} ... B_6^6] in meV.
% E = [E_1 E_2 .. E_2J+1] is a (2J+1)-component vector with the
% known crystal field energies levels in meV
% ind_par = [index_1 ...]
% is a matrix with the index of the parameters to be fitted.
% If ind_par = 0; or not given all non-zero parameters will
% be fitted
% constraints = [15 x 15] matrix of constraints on the CF params.
% The params are indexed as [B20 B21 B22 B40...B44 B60...B66]
% The rows in the constraints matrix indicate the dependent
% parameter and the columns the independent parameter. E.g.
% to specify that B44 = 5*B40, where B44 is the 8th element
% of the parameter vector, and B40 is the 4th, then the
% element constraints(8,4) = 5; To specify B64 = 21*B60,
% the element constraints(13,9) = 21; All other elements is
% zero.
%
% Outputs: FitB2, FitB4, FitB6 are best estimates of the crystal field
% parameters for the given energy levels.
%
% Please note that this function will only attempt to fit the non-zero
% parameters given in B2,B4,B6. If you want a zero initial value for a
% parameter please set it to eps.
%
% This routine is based on the program ENGYFIT.BAS included in the book
% The Crystal Field Handbook by Newman and Ng, CUP 2000.
% By Duc Le (2005) - duc.le@ucl.ac.uk
% The crystal field parameter is given by:
%
% --- q --- q
% > E <l|O |m> > E <l|O |m>
% q ---l,m m k ---l,m m k
% B = ________________________ = ____________________________
% k --- q q q q T
% > <l|O |m> <m|O |l> Tr( <l|O |m> [<m|O |l>] )
% ---l,m k k k k
% To make the equations look nicer
L = A(1); S = A(2); J = A(3);
% The matrix elements of the Stevens operator given by:
% ____________
% k J-M_j -k | (2J+k+1)!
% <LSJM | O |L'SJ'M'> = (-1) ( J k J' ) * 2 | ---------
% j q j (-Mj q Mj') \| (2J-k)!
%
% where this is the 3-j symbol---^ and reduced matrix element--^
%
% Reference: D.Smith and J.H.M. Thornley, Proc. Phys. Soc., 1966, vol 89, pp779.
% Works out the reduced matrix elements
if 2*J-2>0
RM2 = (1/4) * sqrt( factorial(2*J+2+1) / factorial(2*J-2) );
else
RM2 = 0;
end
if 2*J-4>0
RM4 = (1/16) * sqrt( factorial(2*J+4+1) / factorial(2*J-4) );
else
RM4 = 0;
end
if 2*J-6>0
RM6 = (1/64) * sqrt( factorial(2*J+6+1) / factorial(2*J-6) );
else
RM6 = 0;
end
% Works out the matrix elements <i|O_k^q|j>
ind_i = 0;
for Mj = -J:J
ind_i = ind_i + 1;
ind_j = 0;
for Mjp = -J:J
ind_j = ind_j + 1;
% Rank 2
O_mat_el(ind_i,ind_j,1) = (-1)^(J-Mj) * threej([J 2 J; -Mj 0 Mjp]) * RM2 * 2;
O_mat_el(ind_i,ind_j,2) = (-1)^(J-Mj) * threej([J 2 J; -Mj 1 Mjp]) * RM2 *-sqrt(6);
O_mat_el(ind_i,ind_j,3) = (-1)^(J-Mj) * threej([J 2 J; -Mj 2 Mjp]) * RM2 * 2/sqrt(6);
% Rank 4
O_mat_el(ind_i,ind_j,4) = (-1)^(J-Mj) * threej([J 4 J; -Mj 0 Mjp]) * RM4 * 8;
O_mat_el(ind_i,ind_j,5) = (-1)^(J-Mj) * threej([J 4 J; -Mj 1 Mjp]) * RM4 *-2/sqrt(5);
O_mat_el(ind_i,ind_j,6) = (-1)^(J-Mj) * threej([J 4 J; -Mj 2 Mjp]) * RM4 * 4/sqrt(10);
O_mat_el(ind_i,ind_j,7) = (-1)^(J-Mj) * threej([J 4 J; -Mj 3 Mjp]) * RM4 *-2/sqrt(35);
O_mat_el(ind_i,ind_j,8) = (-1)^(J-Mj) * threej([J 4 J; -Mj 4 Mjp]) * RM4 * 8/sqrt(70);
% Rank 6
O_mat_el(ind_i,ind_j,9) = (-1)^(J-Mj) * threej([J 6 J; -Mj 0 Mjp]) * RM6 * 16;
O_mat_el(ind_i,ind_j,10) = (-1)^(J-Mj) * threej([J 6 J; -Mj 1 Mjp]) * RM6 *-8/sqrt(42);
O_mat_el(ind_i,ind_j,11) = (-1)^(J-Mj) * threej([J 6 J; -Mj 2 Mjp]) * RM6 * 16/sqrt(105);
O_mat_el(ind_i,ind_j,12) = (-1)^(J-Mj) * threej([J 6 J; -Mj 3 Mjp]) * RM6 *-8/sqrt(105);
O_mat_el(ind_i,ind_j,13) = (-1)^(J-Mj) * threej([J 6 J; -Mj 4 Mjp]) * RM6 * 16/3/sqrt(14);
O_mat_el(ind_i,ind_j,14) = (-1)^(J-Mj) * threej([J 6 J; -Mj 5 Mjp]) * RM6 *-8/3/sqrt(77);
O_mat_el(ind_i,ind_j,15) = (-1)^(J-Mj) * threej([J 6 J; -Mj 6 Mjp]) * RM6 * 16/sqrt(231);
end
end
% Arranges energies in ascending order and sets ground state to zero.
E = sort(E) - mean(E);
% Initialises values
FitB2 = B2; FitB4 = B4; FitB6 = B6;
FitB = [B2([3 4 5]) B4([5 6 7 8 9]) B6([7 8 9 10 11 12 13])];
leastsqfit = 0;
% NB: B20 = FitB(1)
% B40 = FitB(4) B43 = FitB(7)
% B60 = FitB(9) B63 = FitB(12) B66 = FitB(15)
% Indexes non-zero parameters to fit
if nargin > 5
ind_par_flag = sum(ind_par);
if ~ind_par_flag
ind_par = [(find(B2)-2) (find(B4)-4+3) (find(B6)-6+8)];
end
else
ind_par = [(find(B2)-2) (find(B4)-4+3) (find(B6)-6+8)];
end
% Starts iterations
for num_iteration = 1:100
Hcf = xtalfld_hmltn_stev(A,FitB2,FitB4,FitB6);
[V, Ecalc] = eig(Hcf);
Ecalc = Ecalc(logical(eye(2*J+1)));
if abs( leastsqfit - sum((Ecalc - E').^2) ) < 1e-7
break
end
leastsqfit = sum((Ecalc - E').^2)
Ei = (Ecalc - min(Ecalc))'
for ind_B = ind_par
numer = 0;
% The numerator = sum_i,j( Ei <j|O_k^q|i> )
for ind_i = 1:(2*J+1)
for ind_j = 1:(2*J+1)
for ind_k = 1:(2*J+1)
numer = numer + V(ind_j, ind_i) * O_mat_el(ind_j,ind_k,ind_B) * V(ind_k,ind_i) * E(ind_i);
end
end
end
% The denominator = Tr(<i|O_k^q|j><j|O_k^q|i>)
denom = trace( O_mat_el(:,:,ind_B)' * O_mat_el(:,:,ind_B) );
FitB(ind_B) = numer / denom;
end
% Updates constraints
if nargin > 6
cstr = constraints * FitB';
cstr_ind = find(cstr);
FitB(cstr_ind) = cstr(cstr_ind);
end
FitB2 = [0 0 FitB([1 2 3])];
FitB4 = [0 0 0 0 FitB([4 5 6 7 8])];
FitB6 = [0 0 0 0 0 0 FitB([9 10 11 12 13 14 15])];
end